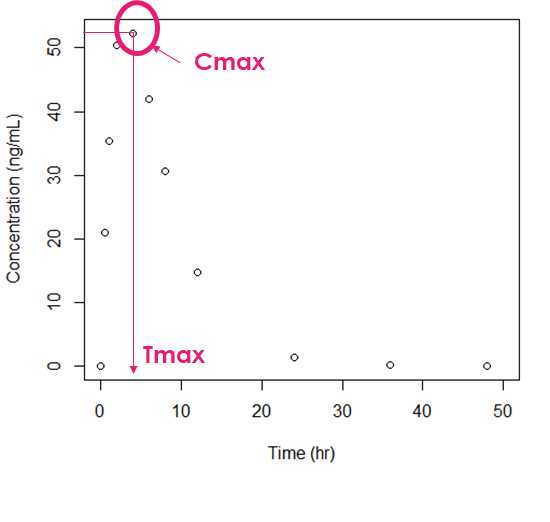
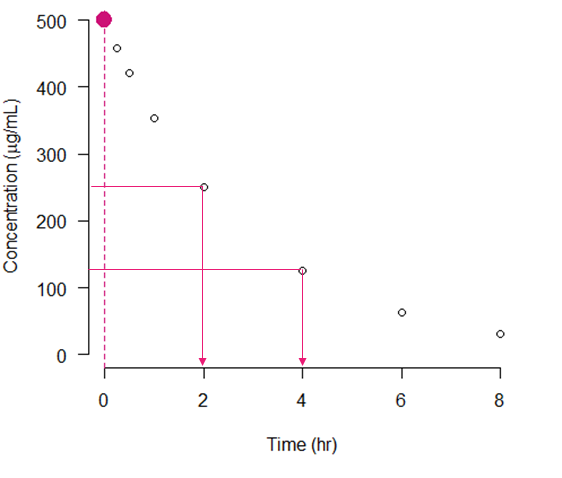
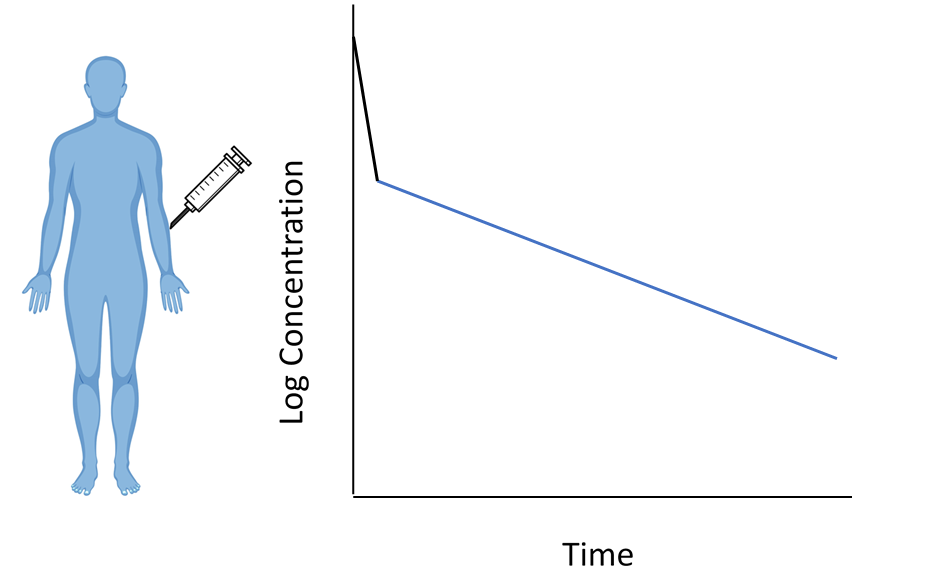
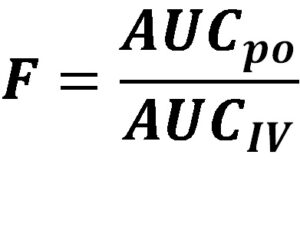Introduction
Pharmacokinetics is the study of how drugs move through the body, including their absorption, distribution, metabolism, and excretion. Among these processes, absorption plays a crucial role in determining the onset, intensity, and duration of a drug’s effects. In this educational blog, we will explore the intricacies of absorption processes, the Biopharmaceutics Classification System (BCS), and how they collectively impact pharmacokinetics.
Absorption: A Gateway to Therapeutic Action
Absorption refers to the movement of a drug from its site of administration into the bloodstream. It is a vital step as it determines the bioavailability of a drug, which is the fraction of the administered dose that reaches systemic circulation unchanged. Understanding the factors that influence absorption is essential for optimizing drug delivery and achieving desired therapeutic outcomes.
Factors Affecting Drug Absorption
1. Route of Administration:
The route by which a drug is administered significantly influences its absorption. Common routes include oral (through the gastrointestinal tract), intravenous (directly into the bloodstream), intramuscular (into the muscle), subcutaneous (under the skin), and topical (applied to the skin). Each route has unique characteristics that impact the rate and extent of absorption.
2. Physicochemical Properties:
The physicochemical properties of a drug, such as molecular weight, solubility, and lipid solubility, play a crucial role in its absorption. Lipophilic (fat-soluble) drugs tend to be absorbed more rapidly than hydrophilic (water-soluble) drugs.
3. Drug Formulation:
The formulation of a drug, including its dosage form and presence of excipients, can affect its absorption. Factors like particle size, pH, and coating can influence drug dissolution and subsequent absorption.
4. Blood Flow:
Blood flow to the site of drug administration determines the rate of absorption. Areas with abundant blood supply, such as the gastrointestinal tract, allow for faster drug absorption compared to areas with limited blood flow.
5. First-Pass Metabolism:
Drugs absorbed through the gastrointestinal tract undergo first-pass metabolism in the liver before reaching systemic circulation. This metabolism can significantly reduce the amount of active drug available, known as the first-pass effect.
Biopharmaceutics Classification System (BCS)
The Biopharmaceutics Classification System (BCS) is a scientific framework that classifies drugs based on their solubility and permeability characteristics. The BCS provides valuable insights into the behavior of drugs in the gastrointestinal tract and their potential for absorption.
The BCS categorizes drugs into four classes (Class I, II, III, and IV) based on their solubility and permeability properties. Let’s explore each class and its implications on drug absorption:
1. Class I:
Class I drugs exhibit high solubility and high permeability. These drugs readily dissolve in the gastrointestinal fluids and easily permeate across the intestinal membranes. They generally exhibit predictable and consistent pharmacokinetics. Examples of Class I drugs include aspirin, acetaminophen, and metoprolol.
2. Class II:
Class II drugs have high permeability but low solubility. Although they can permeate across the intestinal membranes efficiently, their limited solubility can hinder their dissolution in the gastrointestinal fluids. Consequently, the rate and extent of absorption may be variable. Formulation strategies that enhance the solubility of Class II drugs, such as the use of solubilizing agents or particle size reduction techniques, can improve their absorption. Examples of Class II drugs include ketoconazole and ibuprofen.
3. Class III:
Class III drugs have high solubility but low permeability. These drugs readily dissolve
in the gastrointestinal fluids but face challenges in effectively crossing the intestinal membranes. Consequently, their absorption may be limited and variable. Strategies such as prodrug formation or the use of permeation enhancers can be employed to improve their permeability. Examples of Class III drugs include atenolol and cimetidine.
4. Class IV:
Class IV drugs have low solubility and low permeability. These drugs have significant challenges in both dissolution and permeation. They often exhibit poor and inconsistent absorption. Enhancing the solubility and permeability of Class IV drugs is crucial for improving their bioavailability. Examples of Class IV drugs include danazol and griseofulvin.
Impact of Absorption on Pharmacokinetics
1. Onset and Duration of Action:
The rate of drug absorption influences how quickly the drug reaches therapeutic levels in the bloodstream, thereby affecting the onset of action. Additionally, the duration of action depends on the rate at which the drug is absorbed, metabolized, and eliminated from the body.
2. Bioavailability:
The extent of drug absorption determines its bioavailability, which directly affects the dose required to achieve the desired therapeutic effect. High bioavailability indicates efficient absorption, while low bioavailability may necessitate higher doses or alternative routes of administration.
3. Variability in Drug Response:
Variations in drug absorption between individuals can contribute to variability in drug response. Factors such as genetic differences, food intake, disease states, and concomitant drug use can influence drug absorption and lead to variability in pharmacokinetics.
Conclusion
Understanding the absorption processes, including factors affecting drug absorption and the classification of drugs according to the Biopharmaceutics Classification System (BCS), is crucial for optimizing drug therapy. The BCS provides valuable insights into the solubility and permeability characteristics of drugs, guiding formulation strategies for optimal drug delivery. By comprehending these processes, healthcare professionals can make informed decisions regarding dosage, dosing intervals, and route of administration to achieve desired therapeutic outcomes while minimizing the risk of adverse effects.